Bcl2-PPI Inhibitors Library

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
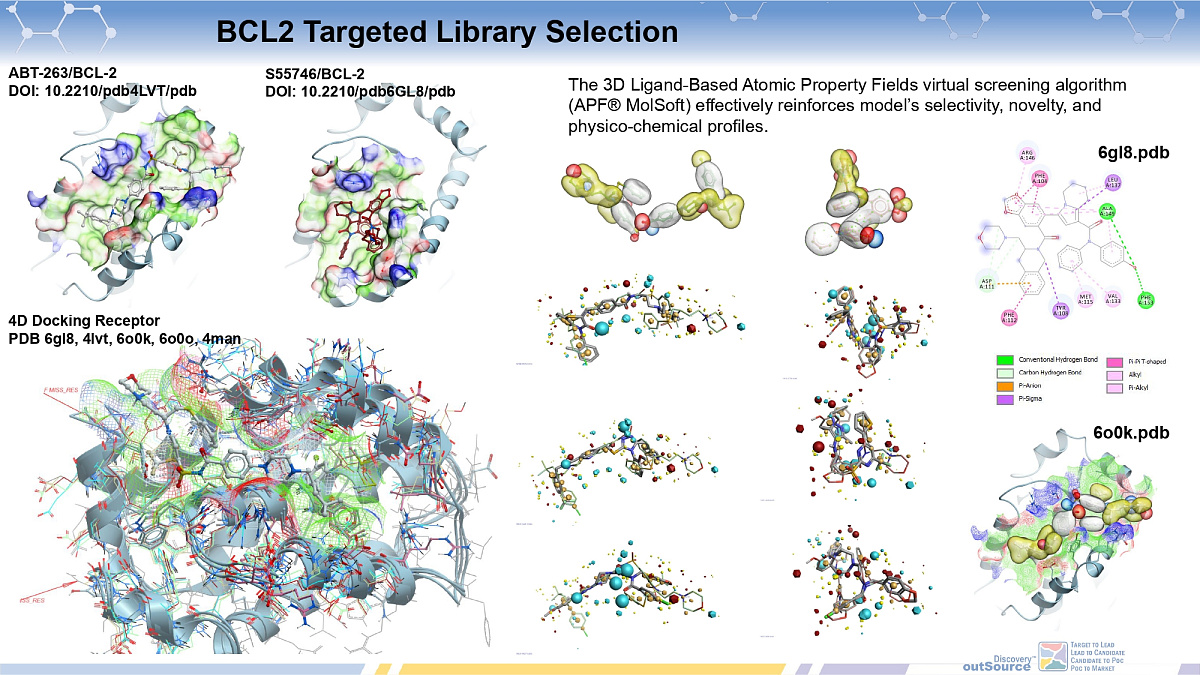
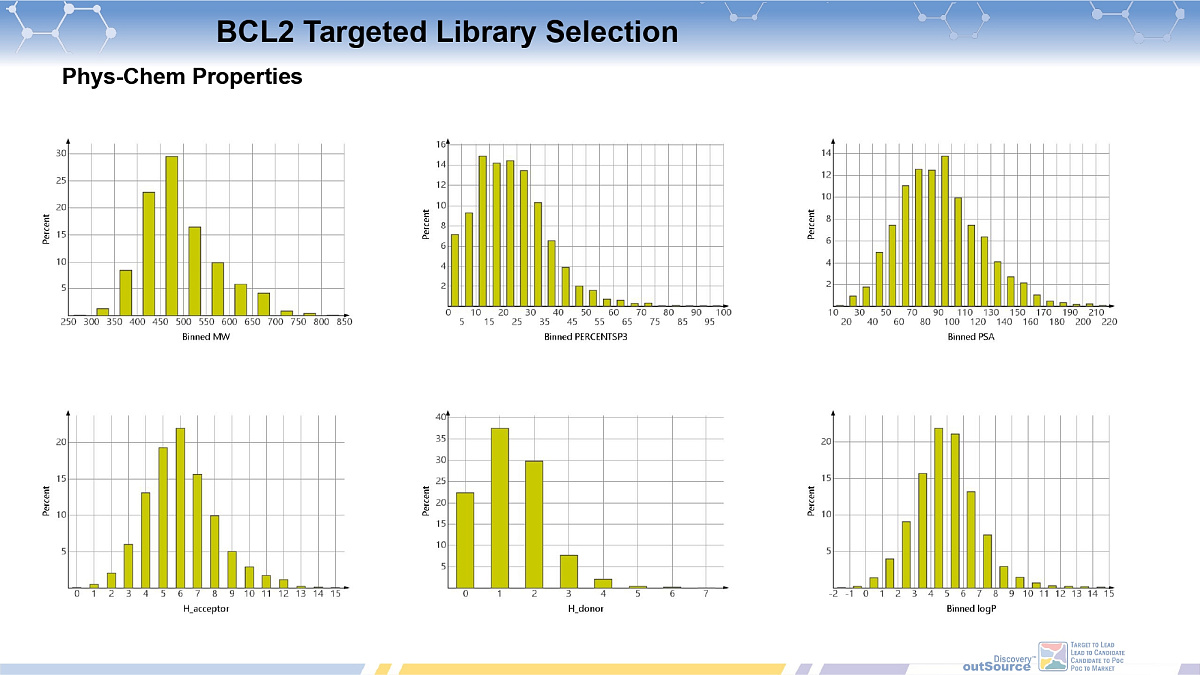
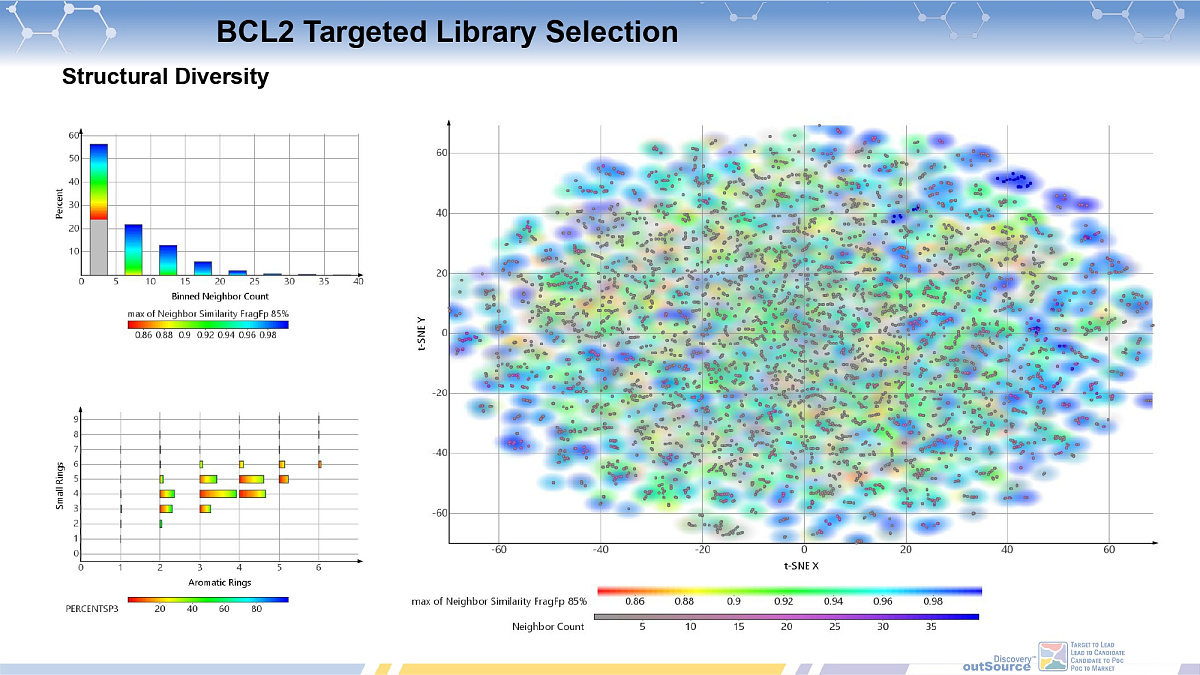
Bcl2-PPI Inhibitors Library
ChemDiv's Bcl-2-PPI Inhibitors Library contains 11,568 compounds.
Proteins in the Bcl-2 family are the central regulators of programmed cell death, with apoptosis-inhibiting members such as Bcl-2 often overexpressed in various cancers, contributing to tumor initiation, progression, and its resistance to therapy. Decreasing Bcl-2 levels has been shown to enhance sensitivity to anticancer drugs and improve in vivo survival outcomes [1]. Currently, two main inhibition strategies for the BCL2 family are being explored in clinical trials. The first strategy involves antisense-based methods to reduce BCL2 expression, while the second employs synthetic BH3 mimetics [2]. Those mimetics represent a novel class of BCL-2 inhibitors that have demonstrated promising results in treating several hematological malignancies, both as standalone treatments and in combination with other anticancer therapies. Their effectiveness in those therapies underscores the potential of targeted BCL2 family inhibition in cancer treatment strategies.
Understanding of the fact that numerous cancers rely on the antiapoptotic functions of Bcl-2 proteins, coupled with the knowledge about their interactions through specific Bcl-2-homology (BH) domains, has spurred the development of drugs that mimic the BH3 domain. These drugs are designed to restore apoptosis by binding to one or more members of the Bcl-2 family. An effective BH3 mimetic should replicate the BH3 domain of a proapoptotic Bcl2 family member, thus inhibiting the antiapoptotic proteins by occupying their BH3-binding groove. There are some successful examples of such inhibitors like ABT-737. It specifically targets the BH3-binding domain of Bcl-2 proteins and demonstrates the ability to induce apoptosis across various cancer models, showcasing its potential as a targeted therapeutic agent in cancer treatment [3].
Bcl2-protein-protein interaction (PPI) inhibitors are the novel agents for drug discovery, particularly in the field of oncology, due to their ability to modulate apoptosis, a key process in cancer progression. By specifically targeting the Bcl2 family of proteins, these inhibitors disrupt the interaction between pro-apoptotic and anti-apoptotic members, thereby reinstating the apoptotic pathway often suppressed in cancer cells. This reinstatement of apoptosis is especially important in treating cancers where the anti-apoptotic Bcl2 proteins are overexpressed, contributing to tumor growth and resistance to conventional therapies. The development of Bcl2-PPI inhibitors, like BH3 mimetics, has provided a novel approach to cancer treatment, offering a strategy to selectively trigger cell death in cancerous cells while sparing healthy cells. Their ongoing research and development continue to expand therapeutic options in cancer, with several Bcl2-PPI inhibitors showing promising results in clinical trials for various malignancies.
Extended collection of the selective Bcl2-PPI inhibitors can help streamline the discovery and development of the novel antitumor therapeutics through the generation and optimization of a series of chemical distinct lead molecules.
References
[1] T. Oltersdorf et al., “An inhibitor of Bcl-2 family proteins induces regression of solid tumours,” Nature, vol. 435, no. 7042, pp. 677–681, 2005, doi: 10.1038/nature03579.
[2] L. Scarfò and P. Ghia, “Reprogramming cell death: BCL2 family inhibition in hematological malignancies,” Immunol. Lett., vol. 155, no. 1–2, pp. 36–39, 2013, doi:
10.1016/j.imlet.2013.09.015.
[3] R. S. Soderquist and A. Eastman, “BCL2 inhibitors as anticancer drugs: A plethora of misleading BH3 mimetics,” Mol. Cancer Ther., vol. 15, no. 9, pp. 2011–2017, 2016, doi: 10.1158/1535-7163.MCT-16-0031.