重点图书馆
基于计算机的聚焦库创建方法。
硅脑
如今,在silico药物设计(CADD)中,绝大多数制药领导者都在使用,包括ChemDiv。 在这里,我们将给出三个使用CADD创建重点库的例子。
循环神经网络(Rnn)
快速发展的CADD方法之一涉及Rnn,在训练后,网络生成类似于输入的输出,即模仿学习数据集中的分子结构的新分子结构。
在文章[1]中,通过转移学习创建自动聚焦库–即首先在一大组(分子,在这种情况下,但概念不限于它们)上进行训练,然后用较小的样本进行优化以进行导
在使用ChEMBL数据集训练RNN之后,选择了模仿药物化学工作流程中通常发生的转移集。
在所有选择的指标中,有两个是评估网络性能的关键:独特新颖性评分和化学接近性评分。
有些违反直觉的是,较小的数据集需要更多的训练,而较大的数据集可以在更少的周期内完成。 较低的片段计数(意味着数据集中较少的不同组-片段-)导致较低的唯一性,因为训练数据更加均匀。
这些结果在这个表中显示(标题数字是指完成的周期(即周期),单元格是指有多少输出(以百分比计)具有低(少于四分之一)独特新颖性得分:
| Filename | Frag count | 5 | 10 | 12 | 15 | 17 | 20 |
| DHODH full | 66 | -- | 1 | 59 | 91 | 96 | 100 |
| METAP2 full | 59 | -- | 60 | 78 | 88 | 91 | 100 |
| MMP-12 full | 31 | 33 | 66 | 80 | 94 | 99 | 100 |
| P2X7 full | 131 | -- | -- | -- | 18 | 78 | 99 |
| SLC22A12 full | 49 | -- | 75 | 83 | 98 | 100 | 100 |
| DHODH subset | 41 | -- | 46 | 62 | 88 | 98 | 100 |
| METAP2 subset | 40 | -- | 60 | 76 | 92 | 100 | 100 |
| MMP-12 subset | 22 | 50 | 80 | 87 | 97 | 100 | 100 |
| P2X7 subset | 64 | -- | 34 | 85 | 95 | 99 | 100 |
| SLC22A12 subset | 32 | 13 | 75 | 88 | 100 | 100 | 100 |
| US-20090018134 | 33 | 8 | 58 | 79 | 91 | 93 | 99 |
| US-20090286778 | 123 | -- | 21 | 55 | 75 | 81 | 83 |
| US-20100016279 | 73 | -- | 82 | 97 | 99 | 100 | 100 |
| US-20120157425 | 91 | 1 | 85 | 92 | 99 | 100 | 100 |
| WO-2010079443 | 54 | -- | -- | -- | 8 | 60 | 92 |
| WO-2011075515 | 137 | -- | 2 | 42 | 89 | 93 | 100 |
| WO-2012053186 | 44 | 1 | 66 | 87 | 94 | 100 | 100 |
| WO-2012067965 | 110 | -- | 34 | 85 | 97 | 98 | 100 |
筛分,筛分
CADD中的另一种常用技术是SBF(基于结构的聚焦),其中使用特定的相互作用约束作为基础来设计可能与目标结合的新化合物。
在文章[2]中,研究人员开发了一种大规模数据分析和可视化的方法–结构信息指纹(sift)。 为了更有效地利用分子的三维性质,开发了r-SIFt,'r'指的是不同的R基团。
在组装虚拟库和对接姿势之后,通过Pipeline Pilot找到二维描述符,此时生成r-SIFts,将绑定参数集成到指纹中。 对于Cscores最高的10个姿势(对于MAP激酶p38抑制剂),随后产生r-SIFts,通过计算Tanimoto系数选择最佳姿势。
通过测量使用先前产生的r-SIFTs做出的决策树的预测精度来评估结果。
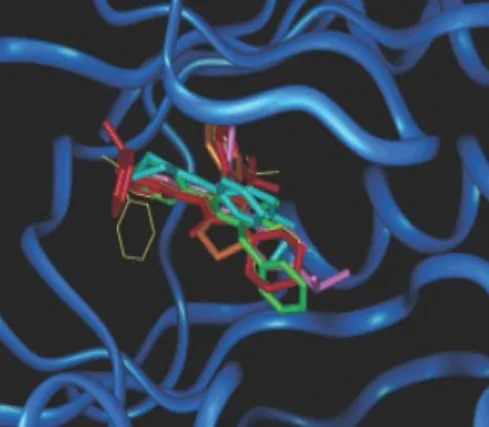
与传统的工具包相结合,r-SIFt被证明是一个很好的可视化工具,可以放大分子的特定部分。 下图显示了p38抑制剂相似的方式,并在进一步检查时揭示了差异。

b是最佳对接姿势的叠加(c-f是p38抑制剂,g不是)。 C的共晶体结构用黄线示出。 抑制剂以类似的方式结合:紫色部分靠近铰链,蓝色部分集中在疏水性口袋中。

结构和R基团。 1-5对应于上图中的c-g。
即,1的R2(紫色c)与铰链的接触比其他更多,这与以前的一致
调查结果。 与较小的3-氟苯酚R1相比,1的三氟苯R1解释了疏水区域中更高程度的相互作用。
多目标遗传算法
多目标遗传算法(MOGA)被用作MoSELECT的基础–一个在虚拟空间中搜索解决方案并呈现不同目标之间的连接的程序[3]。
具有许多目标的任务通常有不同的解决方案,每个解决方案都有不同的权衡。 一个标准的遗传算法分别搜索这些线,不像MOGA,它同时这样做,利用'支配'的想法:

任务是最小化f1和f2。 实心圆用于非主导答案,这意味着对于这两个目标没有更好的解决方案。 空点占主导地位,数字显示有多少"支配者"–更好的解决方案–存在。
当任务是为2-氨基噻唑文库中的随机分子创建一个聚焦文库,优化相似性(通过日光指纹和Tanimoto系数测量)和成本时,SELECT–使用标准遗传算法–只提供了一个单边足够的解决方案–平均值为0,832;US4 48 289,4或0,696;1 675,2。 实现妥协的唯一方法-煞费苦心地选择权重-对于这种不相称的目标来说很难。 MoSELECT,而不是给出单一的解决方案,创建非主导的答案的整个家庭,并允许在决定妥协更容易的选择:

第三个图的扩展版本显示了整个解决方案系列。
结论
总之,在制药行业,硅技术是一个非常有价值的工具。
ChemDiv在化学信息学领域提供一流的CADD服务,包括虚拟筛选、对接、hit2lead优化等。
文学作品
[1]基于RNN转移学习的聚焦文库分子生成指南;Amabilino等。,化学信息与建模杂志2020,60,12,5699-5713
[2]基于知识的基于蛋白质-配体相互作用的靶标文库设计