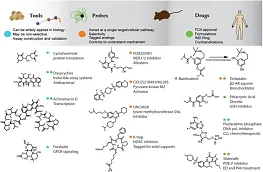
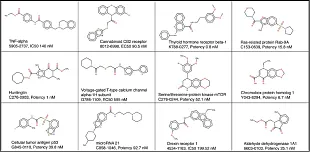
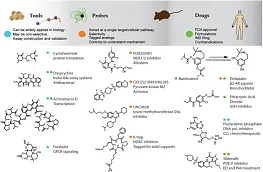
ADME

ADME: fundamentos comerciales
Pilares
Cuatro componentes principales que controlan lo que le sucede a la píldora, o a cualquier otro medicamento, después de que se toman. es decir, la farmacocinética, que no debe confundirse con la farmacodinámica, que describe el otro carril de interacción: lo que el fármaco le hace al paciente. Son absorción, distribución, metabolismo y excreción (ADME para abreviar). En este breve artículo nos centraremos en los conceptos básicos, como introducción para profesionales de otros campos.
Influencias fisicoquímicas
Antes de hablar de ADME en sí, tenemos que discutir brevemente algunos conceptos físicos y químicos, los que tienen un gran impacto en ADME. Para ello, pasaremos por la lipofilia, los enlaces de hidrógeno, el área de superficie polar y la famosa e infame regla de cinco.
Lipofilia
Es de conocimiento común que el aceite y el agua no se mezclan, por lo que se espera que la lipofilia de la molécula, medida por un coeficiente de partición P (simplemente la relación de concentraciones de equilibrio en 1-octanol y agua), afecte todo, desde la solubilidad hasta el aclaramiento. Por conveniencia, log P se escribe normalmente en su lugar.
Si la partición es alta (la molécula es más soluble en 1-octanol), aumenta la unión del receptor y la enzima, al tiempo que disminuye su solubilidad en agua. Además, tales moléculas tienden a unirse mejor a las enzimas CYP450, aumentando así la posibilidad de interacciones fármaco-fármaco. También afecta la unión a proteínas plasmáticas.
Enlaces de hidrógeno
Los enlaces de hidrógeno entre un hidrógeno y un átomo electronegativo como el oxígeno o el nitrógeno pueden formarse dentro de una sola molécula o entre diferentes moléculas. Especialmente importantes son los enlaces de hidrógeno entre el fármaco y las moléculas de agua, porque deben romperse antes de formar otras nuevas, por lo que las sustancias con demasiados enlaces de hidrógeno tienen problemas para abrirse camino desde el tracto gastrointestinal hacia el torrente sanguíneo.
Área de superficie polar
El tamaño de la molécula, por supuesto, también es un factor significativo. Desafortunadamente, es difícil de medir, por lo que se utilizan en su lugar el peso molecular, la densidad electrónica y el área de superficie polar (PSA). PSA es la superficie total de los átomos polares superficiales. El PSA es la métrica de absorción de fármacos más impactante: si la molécula se transporta a través de las células, el límite superior del tamaño es de 120 angstroms cuadrados; y para los fármacos del sistema nervioso central, el límite es aún más bajo, solo 70 angstroms cuadrados.
Regla de cinco
Lipinski y otros determinaron que los fármacos son (la mayoría de ellos, al menos) pequeños y lipófilos, por lo que se debe tener cuidado con los candidatos a fármacos pesados (más de 500 daltons) con un log P grande (más de 5) y demasiados aceptores de enlaces de hidrógeno (más de diez) o donantes (cinco). Debido a su naturaleza directa, esta regla se emplea ampliamente durante el descubrimiento de fármacos.
Sin embargo, hay medicamentos más allá de estas reglas y su participación en el mercado es bastante alta para los medicamentos modernos, por lo que esto no es una garantía, más una guía realmente buena.
En una nota ligeramente diferente, el 95% de los medicamentos no atraviesan la barrera hematoencefálica. Por lo general, una mayor capacidad de enlace de hidrógeno es un problema. Lipinski también tiene una regla para los medicamentos destinados al uso en el sistema nervioso central: peso aún menor que antes (400 dalton), log P menor que 5, incluso menos donantes de enlaces de hidrógeno (menos de cuatro) y aceptores (menos de ocho).
A significa Absorción
Hay tres fases en el viaje de la medicina oral a través del paciente: fase de absorción (siendo la absorción el proceso dominante), postabsorción (prevalece la eliminación), eliminación (sin absorción en esta etapa).
Los parámetros convencionales para este viaje (figura 1) son: Cmax, la concentración máxima o concentración máxima; tmax, el tiempo para alcanzar Cmax; Cmin, la concentración mínima observada y el área AUC bajo la curva, que corresponde a la biodisponibilidad. Otro parámetro, más abstracto que el físico, es el volumen de distribución Vd – dosis dividido por la concentración. La dosis es solo la cantidad total de fármaco, mientras que la concentración es específicamente sobre el plasma. El aclaramiento se refiere al volumen de sangre limpiado por unidad de tiempo. Por último, está la vida media: 0,693 veces el volumen de distribución dividido por el aclaramiento.
La segunda figura muestra la absorción en un solo esquema, destacando el hecho de que los medicamentos terminan pasando por el hígado antes de convertirse en parte del torrente sanguíneo general, por lo que siempre se deben tener en cuenta las formas en que el hígado interactúa con el medicamento.

Figure 1. A standard concentration-time graph for oral drugs.
D is for Distribution
After being absorbed, drug is distributed through the bloodstream to the tissues. Distribution denotes the process of the medicine reversibly leaving the blood and going to the tissue. For a sense of scale, plasma only accounts for 5% body mass, while fat, intestinal fluid, intracellular fluid and transcellular fluid account for 20%, 16%, 35%, 2% correspondingly.
Principally, the entirety of body water has three categories: intravascular, interstitial, intracellular. Drugs will be distributed into these three differently depending on their nature and may gather in organs – most notably, in liver or kidneys. And lipophilic drugs will gather in fat tissue.
The entirety of the drug itself is compartmentalized over numerous membranes, tissue, blood and physiological volumes.So if the volume of distribution is extremely high, for example, fifteen thousand liters (assuming the weight of seventy kilograms), one should understand that as the most of the substance concentrated in the tissue.

Figura 1. Un gráfico estándar de concentración-tiempo para fármacos orales.
D es para distribución
Después de ser absorbido, el fármaco se distribuye a través del torrente sanguíneo a los tejidos. La distribución denota el proceso del medicamento que sale de la sangre de manera reversible y va al tejido. Para una sensación de escala, el plasma solo representa el 5% de la masa corporal, mientras que la grasa, el líquido intestinal, el líquido intracelular y el líquido transcelular representan 20%, 16%, 35%, 2% correspondientemente.
Principalmente, la totalidad del agua corporal tiene tres categorías: intravascular, intersticial, intracelular. Los medicamentos se distribuirán en estos tres de manera diferente dependiendo de su naturaleza y pueden acumularse en los órganos, especialmente en el hígado o los riñones. Y los fármacos lipófilos se acumularán en el tejido graso.
La totalidad del fármaco en sí está compartimentada en numerosas membranas, tejidos, sangre y fisiológicos volumes.So si el volumen de distribución es extremadamente alto, por ejemplo, quince mil litros (suponiendo el peso de setenta kilogramos), se debe entender que la mayor parte de la sustancia se concentra en el tejido.
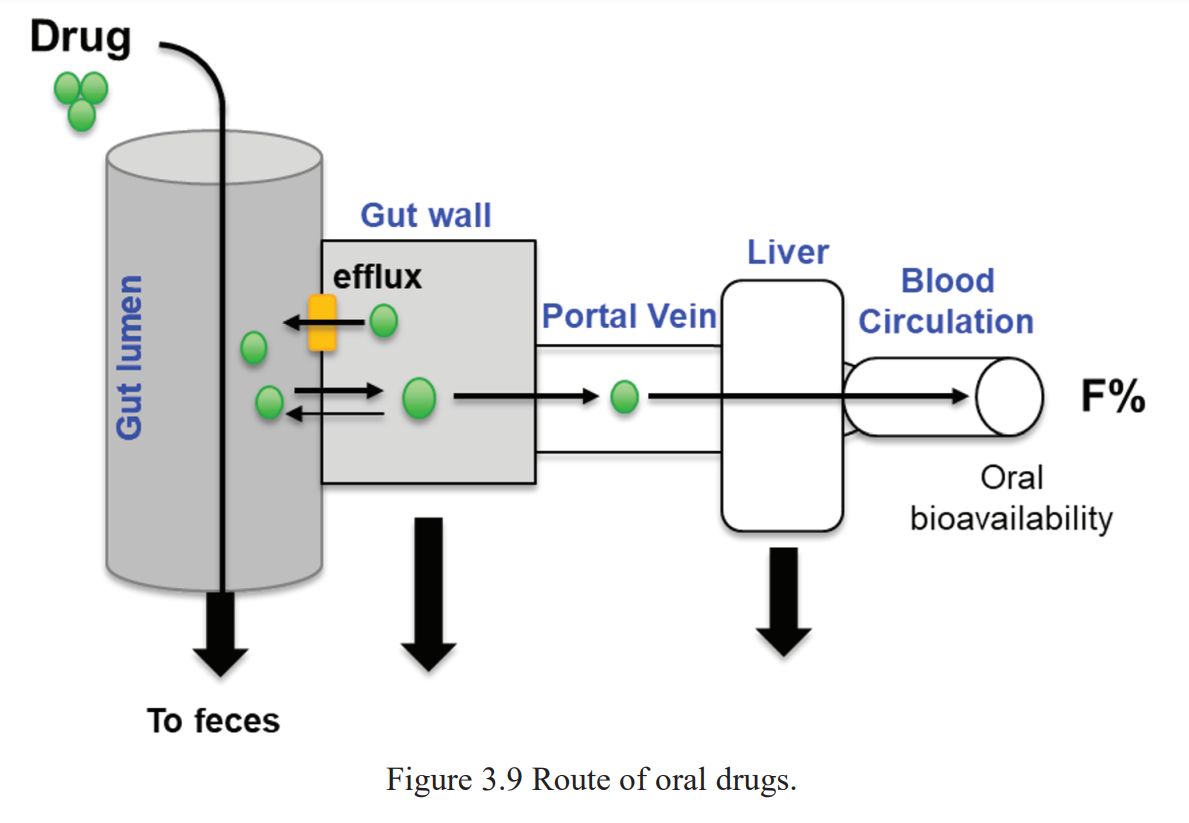
Figura 2. Absorción y distribución temprana de un fármaco, con metabolismo en el hígado;
Metabolismo en el medio
A menos que se metabolicen, los fármacos lipófilos y algunos hidrófilos terminarían acumulándose en el cuerpo. En general, las sustancias lipófilas se convierten en metabolitos hidrófilos, de los que es más fácil deshacerse. El jugador indiscutiblemente principal aquí es el hígado, pero otros sitios (riñones, plasma, intestinos, pulmones) también juegan un papel. Mientras que la mayoría de los compuestos se transforman en metabolitos inactivos, algunos se convierten en activos. De hecho, algunos medicamentos en realidad se convierten en su forma activa en el hígado, y la molécula consumida de antemano es un profármaco.
A nivel molecular, la clase de enzimas CYP450 es en gran parte responsable del metabolismo de los fármacos. De estos, 3A4 hace alrededor de la mitad del trabajo. Otras enzimas de la misma familia son 2D6, 1A2, 2C9, 2C19 - junto con 3A4 manejan el 90% de los fármacos.
Hay dos fases del metabolismo de los fármacos, la primera transforma los grupos funcionales y la segunda consiste en añadir un componente muy hidrófilo a la molécula original o al producto resultante de la primera fase (figura 3).
< img width= " 957 "src="/images/3.png" height="710">
Figura 3. Dos fases del metabolismo
El metabolismo de la fase uno generalmente se puede describir como un proceso de oxidación: desde la hidroxilación hasta la oxigenación en átomos de azufre. Estas reacciones se realizan en gran medida por monooxigenasas hepáticas.
El metabolismo de la segunda fase consiste en la conjugación con un sustrato para formar una sustancia fácilmente eliminable.
Termina con excreción
Después de lidiar con todo eso, es hora de deshacerse de la droga, mediante la eliminación de la excreción. La mayoría de las veces, pasan por la vía renal y terminan en la orina. Cuando se trata de cinética, hay no lineales (de primer orden) y lineales (de orden cero). La mayoría de los fármacos siguen una cinética no lineal, por lo que la fracción constante se extrae cada hora, lo que significa que la tasa de eliminación depende de la concentración. Para cinética lineal, se elimina la misma cantidad de fármaco cada hora. Cualquier fármaco en alta concentración puede comportarse de esa manera.
La vía renal tiene tres partes: filtración glomerular, reabsorción tubular y secreción tubular.