For 30 years, drug companies poured resources into the same hypothesis: clear the beta-amyloid protein from the brain, and Alzheimer's would slow. More than 200 clinical trials later, no treatment has successfully halted the disease. In July 2022, Science magazine reported that a pivotal 2006 Nature study underpinning that hypothesis may have been based on falsified data.
The Failure of the Amyloid Cascade Hypothesis
Beta-amyloid became the central target of Alzheimer’s research after a 1992 paper in Nature proposed what became known as the amyloid cascade hypothesis, that beta-amyloid accumulation triggers the disease. Subsequent research piled up supporting the theory. Drug development followed. Yet every major anti-amyloid therapy either failed in clinical trials or, like aducanumab (Aduhelm), received controversial FDA approval in June 2021 before being withdrawn from the market entirely in January 2024 amid safety and efficacy concerns.
Over 200 clinical trials across three decades produced no disease modifying treatment. Though beta-amyloid plaques are present in Alzheimer’s brains, clearing them wasn’t producing the outcomes the hypothesis predicted.
A Different Model Entirely: Alzheimer's as an Autoimmune Disease
Dr. Donald Weaver, Director of the Krembil Brain Institute and Professor of Chemistry, Medicine and Pharmaceutical Sciences at the University of Toronto, has spent more than 30 years developing an alternative explanation. His model, published in Alzheimer’s & Dementia and recognized with the Oskar Fischer Prize from the University of Texas at San Antonio, reframes the disease entirely.
“We don’t think of Alzheimer’s as fundamentally a disease of the brain,” Weaver told the University Health Network. “We think of it as a disease of the immune system within the brain.”
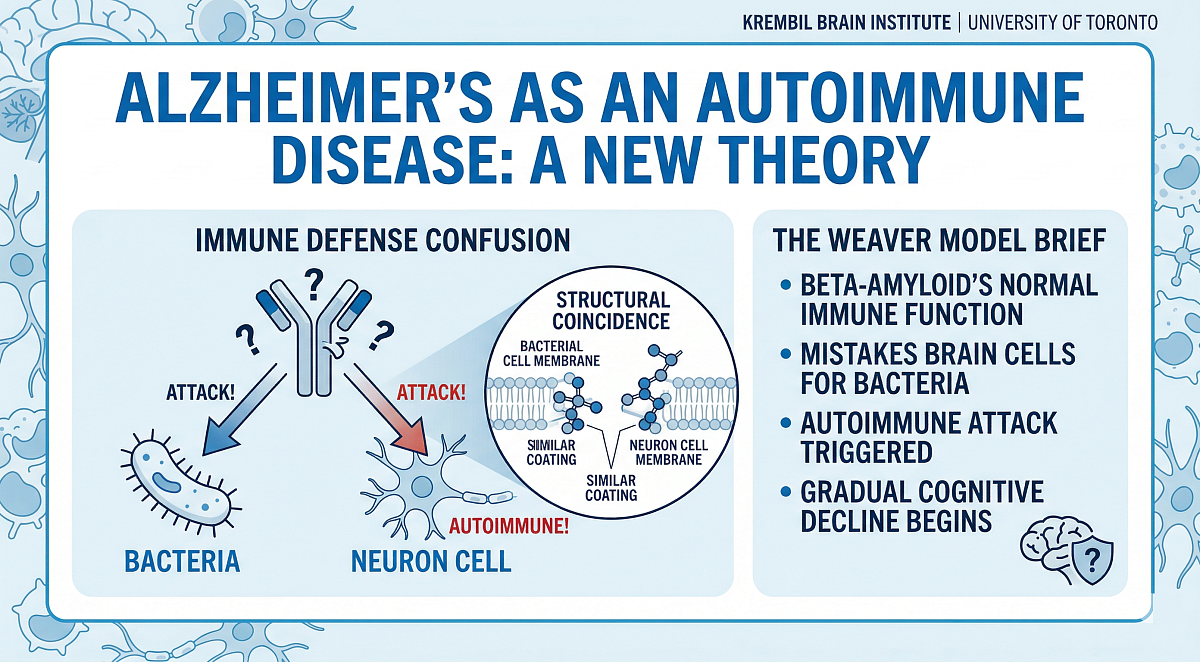
In Weaver’s model, beta-amyloid is not an abnormal protein. It is a normal component of the brain’s innate immune system. Considered to be present by design, released in response to head trauma, bacterial infection, pollution, or other threats. The problem arises from a structural coincidence. The fat molecules forming bacterial cell membranes are similar to those forming the membranes of brain cells. So much so that beta-amyloid cannot distinguish between the two. In attacking the bacteria it was deployed to destroy, it simultaneously attacks the neurons it was supposed to protect.
“Beta-amyloid gets confused,” Weaver explained. “It can’t tell the difference between a bacteria and a brain cell and so it inadvertently attacks our own brain cells. This becomes what we call an autoimmune disease — the immune system attacking the host.” When beta-amyloid destroys a neuron, the breakdown products trigger further beta-amyloid release, creating a self-perpetuating cycle that gradually escalates over years into the cognitive decline seen in Alzheimer’s patients.
Why Conventional Autoimmune Treatments Won’t Work
The autoimmune framing raises an obvious question: why not treat Alzheimer’s the way medicine treats other autoimmune diseases? Specifically, rheumatoid arthritis, lupus, multiple sclerosis with steroid based or immunosuppressive therapies. Weaver points out that the brain is not like other organs. Systemic immunosuppression that works in joints or connective tissue would devastate the neural immune system that Alzheimer’s patients need functioning.
Instead, his team has screened approximately 1,300 chemicals in the brain for their ability to selectively modulate specific immune pathways. A second paper, published in Alzheimer’s & Dementia: Translational Research & Clinical Interventions, identified tryptophan metabolites. These are biological products of the amino acid tryptophan, particularly effective at preventing beta-amyloid accumulation. These compounds now form the basis of a therapeutic pipeline targeting the autoimmune mechanism rather than the plaques it produces.
Other Emerging Theories
Weaver’s model is not the only challenge to the amyloid hypothesis gaining traction. Some researchers now classify Alzheimer’s as a mitochondrial disease, a failure of the energy generating structures inside brain cells. Others point to bacterial infection, particularly oral bacteria, as a trigger. A third group investigates abnormal handling of metals including zinc, copper, and iron in brain tissue.
Each theory approaches the same disease through a different lens. What they share is the recognition that beta-amyloid accumulation may be a symptom rather than a cause, a downstream consequence of processes the amyloid hypothesis never fully explained.
More than 50 million people worldwide currently live with dementia. A new diagnosis occurs every three seconds. The amyloid hypothesis dominated research for 30 years and produced no cure. Weaver’s model doesn’t promise one either but it asks a different question, and after three decades of the same answer, that may be exactly what the field needs.