Lipid Metabolism Library

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
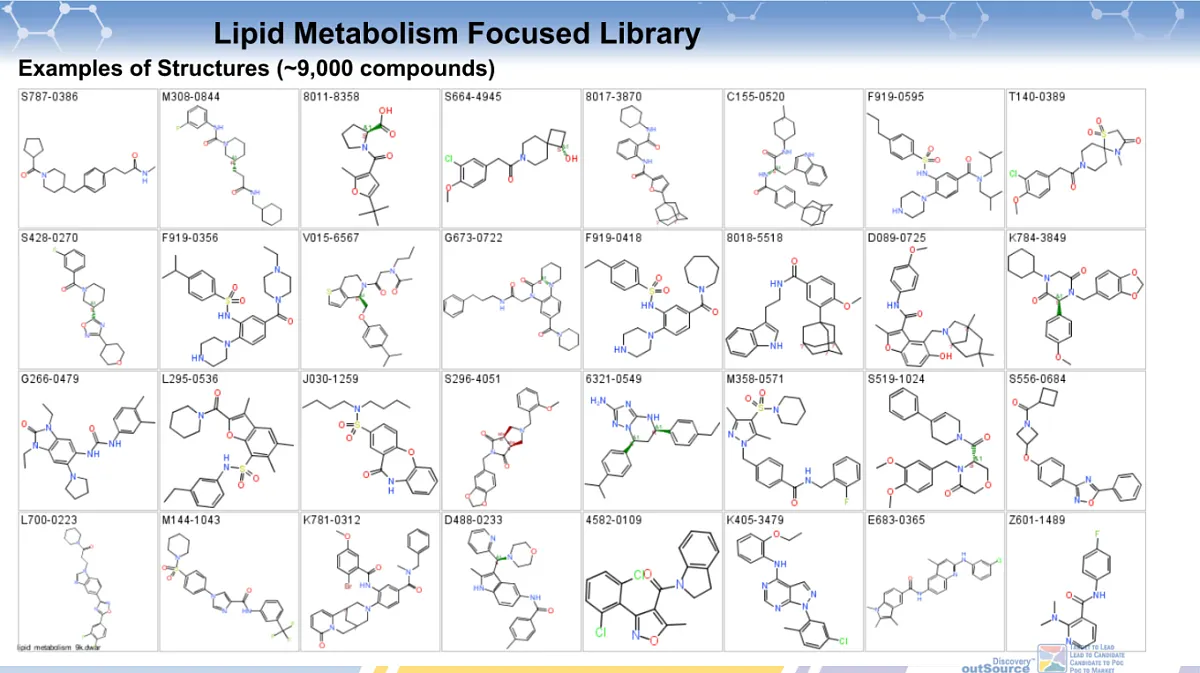
The ChemDiv’s library of small molecules targeting pathways involved in lipid metabolism contains 9,149 compounds.
Lipid metabolism disorders, encompassing inherited errors of lipid metabolism, represent a spectrum of conditions characterized by disturbances in the catabolism and synthesis of lipids and lipid-like substances. These disorders are commonly associated with elevated plasma lipid concentrations, including LDL cholesterol, VLDL, and triglycerides. Such elevations are significant risk factors for the development of cardiovascular diseases. These conditions not only involve complexities in lipid breakdown but also in their biosynthesis, underscoring the critical balance required in lipid homeostasis for maintaining cardiovascular health.
The ChemDiv virtual screening methodology included several steps:
- Swiss-Prot Protein Targets and PDB X-Ray Structure Search: This step involves identifying relevant protein targets using the Swiss-Prot database and searching for X-ray crystal structures of these proteins in the Protein Data Bank (PDB).
- Training Sets Compilation: Utilizes data from ChEMBL 25, PubMed, and current patent literature (including sources like CAS and Integrity) to create comprehensive training sets for machine learning models.
- Machine Learning Data Curation:
- Utilizes tools like KNIME/RDKit for data preprocessing and implements a k-Nearest Neighbors (kNN) classifier. This step involves measuring distance in BitVector Cosine Space and using FCFP12 (10,240 bit) fingerprints for analysis.
- Develops a Hybrid 2D QSAR/Fingerprint Model using Kernel Chemical Classification/Regression (kcc).
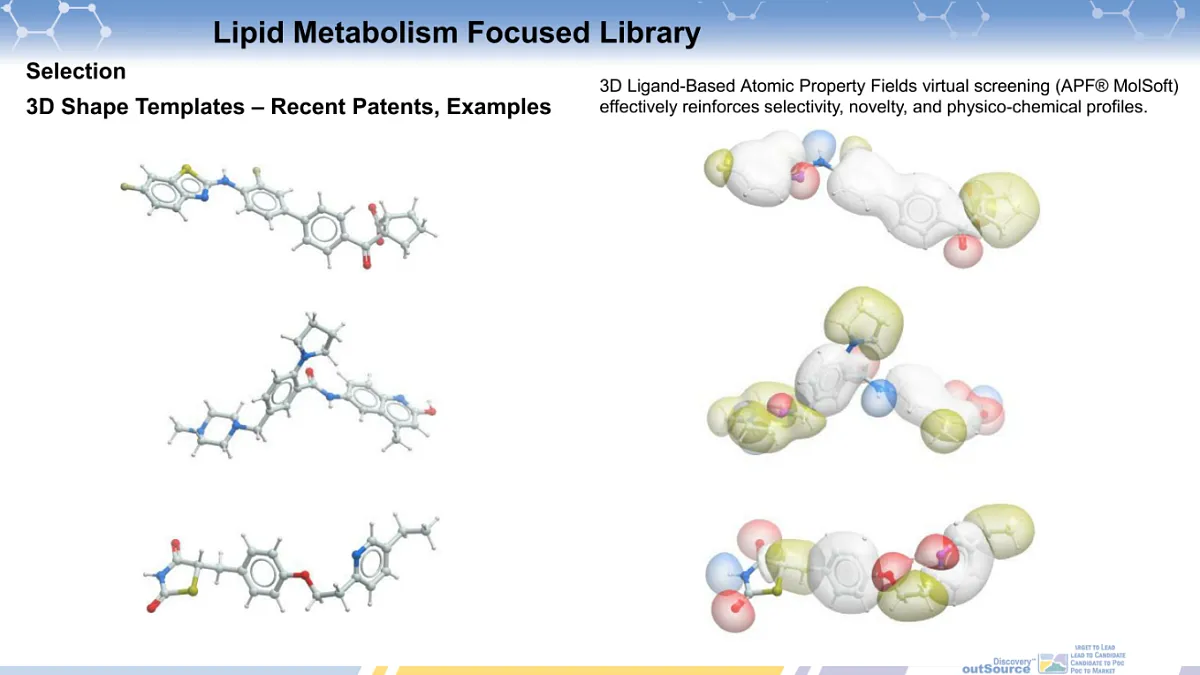
- 3D Shape Similarity Virtual Screening: Employs APF® (Atom-Property Field) from MolSoft and references Lam et al.’s work published in the Journal of Computer-Aided Molecular Design in 2018 and 2019 [1-3], as well as article by Totrov published in Chemical Biology & Drug Design in 2008 [4] for 3D shape similarity assessments.
- Structure-Based Docking / Virtual Screening:
- Implements Multiple Receptor Conformation (MRC) 4D Docking using ICM-Pro from MolSoft, referencing Bottegoni et al. (2009) in the Journal of Medicinal Chemistry [5].
- Uses Ligand-Biased Ensemble receptor Docking (LigBEnD) technique with ICM-Pro from MolSoft, as described by Lam et al. in the Journal of Computer-Aided Molecular Design [1].
- REOS, MedChem & PAINS Filters: This step involves the removal of reactive, toxic, promiscuous, and other undesirable structural motifs from the data sets, ensuring the selection of high-quality compounds.
- Diversity Picking (Tanimoto): Implements the RDKit version of the MaxMin algorithm, based on Ashton, M. et al.’s work in Quantitative Structure-Activity Relationships in 2002 [6], to select diverse compounds for further investigation.
Each of these steps contributes to a robust and comprehensive virtual screening process, leveraging a combination of database searches, machine learning, molecular docking, and compound filtering to identify potential drug candidates efficiently.
This library, meticulously curated through advanced virtual screening techniques and enriched with diverse chemical entities, serves as a vital resource for identifying potential drug candidates. By incorporating a vast array of compounds, each with unique structural and pharmacological properties, the library allows rapid and efficient testing of a multitude of interactions against a variety of biological targets involved lipid metabolism regulation. This approach significantly accelerates the initial phase of drug discovery by swiftly narrowing down the vast chemical space to a more manageable set of promising molecules. Hence, it enhances the likelihood of identifying effective therapeutic agents and reduces time and resources typically expended in the early stages of drug development. The use of our comprehensive and well-structured screening library helps the integration of innovative computational tools and empirical data, ultimately paving the way for more efficient and targeted drug discovery stages.
By providing a rich repository of compounds to test against specific biological targets related to lipid metabolism, the library significantly streamlines the identification of viable drug candidates. This targeted approach not only accelerates the initial phase of discovering lipid-regulating drugs but also increases the likelihood of uncovering effective therapeutics that can address disorders of lipid metabolism. The use of such an expansive and meticulously curated screening library underscores the synergy between cutting-edge computational strategies and empirical research, driving forward the development of specialized treatments in the realm of lipid metabolism and associated diseases.
References
[1] Lam PC, Abagyan R, Totrov M. Hybrid receptor structure/ligand-based docking and activity prediction in ICM: development and evaluation in D3R Grand Challenge 3. J Comput Aided Mol Des. 2019 Jan;33(1):35-46. doi: 10.1007/s10822-018-0139-5
[2] Lam PC, Abagyan R, Totrov M. Macrocycle modeling in ICM: benchmarking and evaluation in D3R Grand Challenge 4. J Comput Aided Mol Des. 2019 Dec;33(12):1057-1069. doi: 10.1007/s10822-019-00225-9.
[3] Lam PC, Abagyan R, Totrov M. Ligand-biased ensemble receptor docking (LigBEnD): a hybrid ligand/receptor structure-based approach. J Comput Aided Mol Des. 2018 Jan;32(1):187-198. doi: 10.1007/s10822-017-0058-x.
[4] Totrov M. Atomic property fields: generalized 3D pharmacophoric potential for automated ligand superposition, pharmacophore elucidation and 3D QSAR. Chem Biol Drug Des. 2008. Jan;71(1):15-27. doi: 10.1111/j.1747-0285.2007.00605.x.
[5] Bottegoni G, Kufareva I, Totrov M, Abagyan R. Four-dimensional docking: a fast and accurate account of discrete receptor flexibility in ligand docking. J Med Chem. 2009 Jan 22;52(2):397-406. doi: 10.1021/jm8009958.
[6] Ashton, M., Barnard, J., Casset, F., Charlton, M., Downs, G., Gorse, D., Holliday, J., Lahana, R. and Willett, P. (2002), Identification of Diverse Database Subsets using Property-Based and Fragment-Based Molecular Descriptions. Quant. Struct.-Act. Relat., 21: 598-604. https://doi.org/10.1002/qsar.200290002