FDA approves first treatment for certain patients with Erdheim-Chester Disease, a rare blood cancer
The U.S. Food and Drug Administration today expanded the approval of Zelboraf (vemurafenib) to include the treatment of certain adult patients with Erdheim-Chester Disease (ECD), a rare cancer of the blood. Zelboraf is indicated to treat patients whose cancer cells have a specific genetic mutation known as BRAF V600. This is the first FDA-approved treatment for ECD.
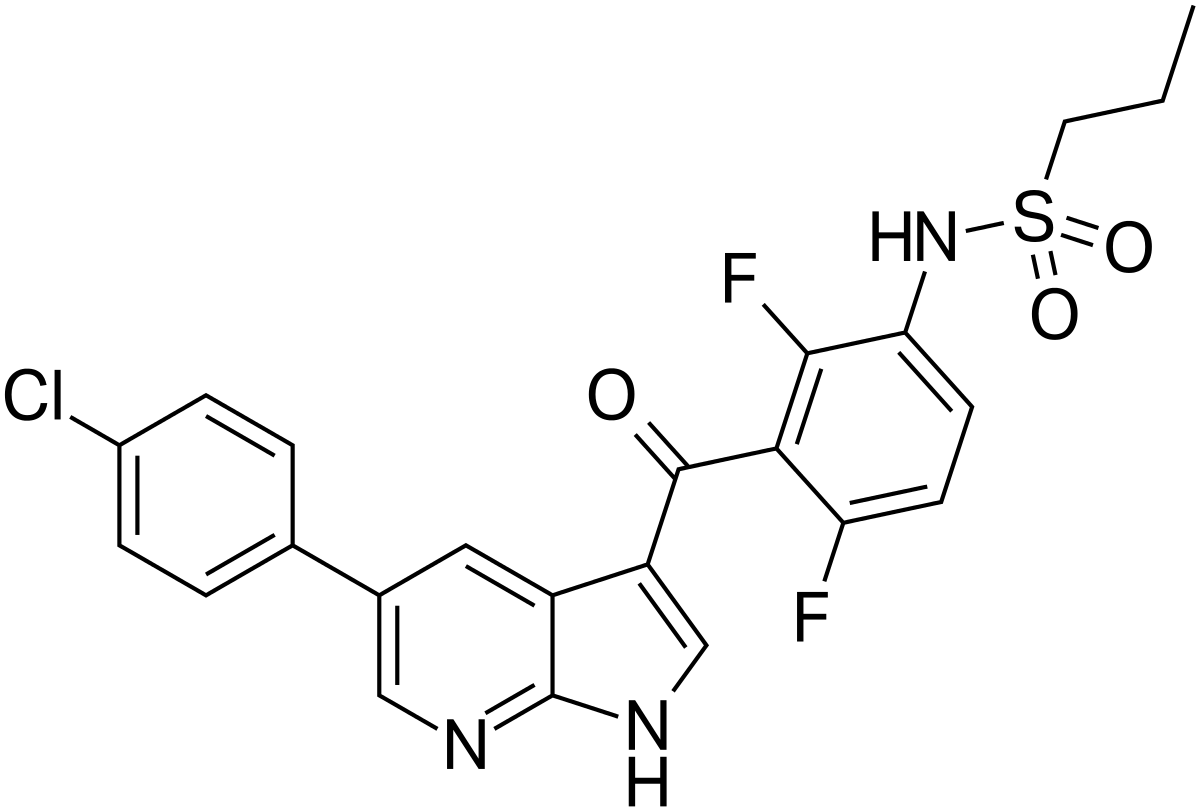
“Today’s approval of Zelboraf for patients with ECD demonstrates how we can apply knowledge of the underlying genetic characteristics of certain malignancies to other cancers,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “This product was first approved in 2011 to treat certain patients with melanoma that harbor the BRAF V600E mutation, and we are now bringing the therapy to patients with a rare cancer with no approved therapies.”
ECD is a slow-growing blood cancer that originates in the bone marrow. ECD causes an increased production of histiocytes, a type of white blood cell. Excess histiocytes can result in tumors infiltrating many organs and tissues throughout the body, including the heart, lungs, brain and others. ECD is estimated to affect 600 to 700 patients worldwide. Approximately 54 percent of patients with ECD have the BRAF V600 mutation. Patients with ECD have very limited life expectancies.
Zelboraf is a kinase inhibitor that works by blocking certain enzymes that promote cell growth.
The efficacy of Zelboraf for the treatment of ECD was studied in 22 patients with BRAF-V600-mutation positive ECD. The trial measured the percent of patients who experienced a complete or partial reduction in tumor size (overall response rate). In the trial, 11 patients (50 percent) experienced a partial response and 1 patient (4.5 percent) experienced a complete response.
Common side effects of Zelboraf in patients with ECD include joint pain (arthralgia); small, raised bumps (maculo-papular rash); hair loss (alopecia); fatigue; change in the heart’s electrical activity (prolonged QT interval) and skin growths (papilloma).
Severe side effects of Zelboraf include the development of new cancers (skin cancer, squamous cell carcinoma or other cancers), growth of tumors in patients with BRAF wild-type melanoma, hypersensitivity reactions (anaphylaxis and DRESS syndrome), severe skin reactions (Stevens-Johnson Syndrome and toxic epidermal necrolysis), heart abnormalities (QT prolongation), liver damage (hepatotoxicity), photosensitivity, severe reactions in the eye (uveitis), immune reactions after receiving radiation treatment (radiation sensitization and radiation recall), kidney failure and thickening of tissue in the hands and feet (Dupuytren’s contracture and plantar fascial fibromatosis). Zelboraf can cause harm to a developing fetus; women should be advised of the potential risk to the fetus and to use effective contraception.
The FDA granted this application Priority Review and Breakthrough Therapy designations for this indication. Zelboraf also received Orphan Drug designation for this indication, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Zelboraf to Hoffman-LaRoche, Inc.
The FDA, an agency within the U.S. Department of Health and Human Services, promotes and protects the public health by, among other things, assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices. The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.