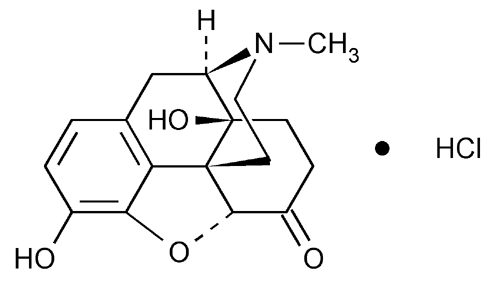
FDA requests removal of Opana ER for risks related to abuse
Today, the U.S. Food and Drug Administration requested that Endo Pharmaceuticals remove its opioid pain medication, reformulated Opana ER (oxymorphone hydrochloride), from the market. After careful consideration, the agency is seeking removal based on its concern that the benefits of the drug may no longer outweigh its risks. This is the first time the agency has taken steps to remove a currently marketed opioid pain medication from sale due to the public health consequences of abuse.
“We are facing an opioid epidemic – a public health crisis, and we must take all necessary steps to reduce the scope of opioid misuse and abuse,” said FDA Commissioner Scott Gottlieb, M.D. “We will continue to take regulatory steps when we see situations where an opioid product’s risks outweigh its benefits, not only for its intended patient population but also in regard to its potential for misuse and abuse.”
The FDA’s decision is based on a review of all available postmarketing data, which demonstrated a significant shift in the route of abuse of Opana ER from nasal to injection following the product’s reformulation. Injection abuse of reformulated Opana ER has been associated with a serious outbreak of HIV and hepatitis C, as well as cases of a serious blood disorder (thrombotic microangiopathy). This decision follows a March 2017 FDA advisory committee meeting where a group of independent experts voted 18-8 that the benefits of reformulated Opana ER no longer outweigh its risks.
Opana ER was first approved in 2006 for the management of moderate-to-severe pain when a continuous, around-the-clock opioid analgesic is needed for an extended period of time. In 2012, Endo replaced the original formulation of Opana ER with a new formulation intended to make the drug resistant to physical and chemical manipulation for abuse by snorting or injecting. While the product met the regulatory standards for approval, the FDA determined that the data did not show that the reformulation could be expected to meaningfully reduce abuse and declined the company’s request to include labeling describing potentially abuse-deterrent properties for Opana ER. Now, with more information about the risks of the reformulated product, the agency is taking steps to remove the reformulated Opana ER from the market.
“The abuse and manipulation of reformulated Opana ER by injection has resulted in a serious disease outbreak. When we determined that the product had dangerous unintended consequences, we made a decision to request its withdrawal from the market,” said Janet Woodcock, M.D., director of the FDA’s Center for Drug Evaluation and Research. “This action will protect the public from further potential for misuse and abuse of this product.”
The FDA has requested that the company voluntarily remove reformulated Opana ER from the market. Should the company choose not to remove the product, the agency intends to take steps to formally require its removal by withdrawing approval. In the interim, the FDA is making health care professionals and others aware of the particularly serious risks associated with the abuse of this product.
The FDA will continue to examine the risk-benefit profile of all approved opioid analgesic products and take further actions as appropriate as a part of our response to this public health crisis.
The FDA, an agency within the U.S. Department of Health and Human Services, promotes and protects the public health by, among other things, assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices. The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.
June 8, 2017
Source: https://www.fda.gov/