KRAS-Targeted Library

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
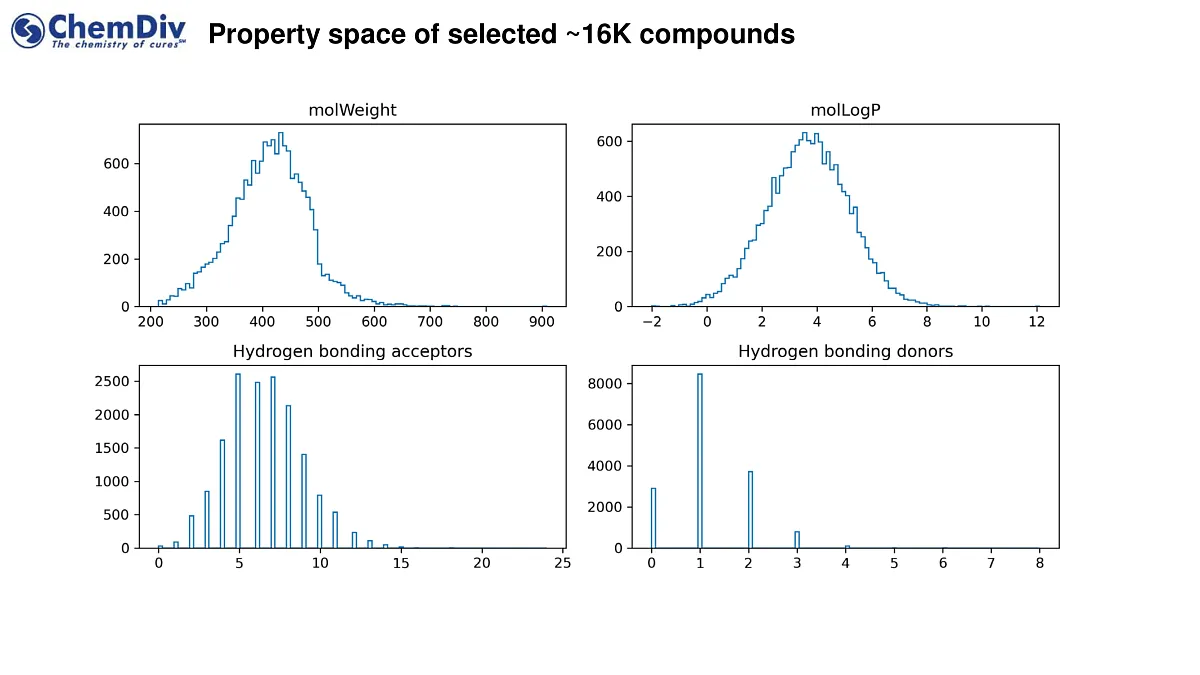
ChemDiv’s Library of the KRAS-Targeting Compounds comprises 16,000 molecules.
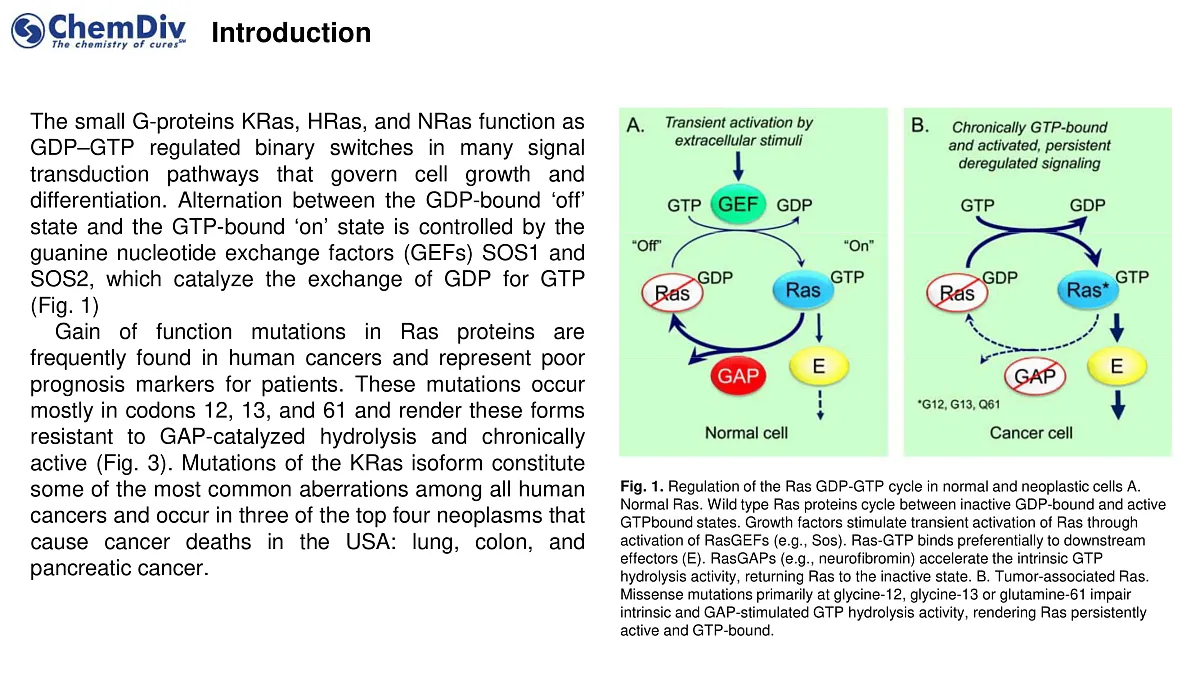
The small G-proteins KRas, HRas, and NRas function as GDP–GTP regulated binary switches in numerous signal transduction pathways that control cell growth and differentiation. The alternation between the GDP-bound ‘off’ state and the GTP-bound ‘on’ state is regulated by guanine nucleotide exchange factors (GEFs) SOS1 and SOS2, which facilitate the exchange of GDP for GTP.
Gain-of-function mutations in Ras proteins are frequently observed in human cancers and are considered poor prognostic markers for patients. These mutations predominantly occur in codons 12, 13, and 61, resulting in forms that are resistant to GAP (GTPase-activating proteins)-catalyzed hydrolysis, thereby remaining chronically active. Mutations in the KRas isoform are among the most common genetic alterations in all human cancers, particularly prevalent in three of the top four neoplasms leading to cancer deaths in the USA: lung, colon, and pancreatic cancer.
KRAS is a pivotal target in cancer therapy as it controls cell growth and differentiation. Mutations in KRAS lead to continuous activation of cell proliferation pathways, contributing significantly to cancer development. Historically deemed "undruggable" because of its structure, recent advancements have led to promising strategies for targeting mutant KRAS proteins. KRAS mutations are crucial in precision medicine, enabling personalized treatment approaches and serving as biomarkers for diagnosing specific cancers and predicting treatment responses. Drugs targeting KRAS often form part of combination therapies, aiming to inhibit multiple pathways or overcome resistance mechanisms in cancer treatment. Consequently, KRAS is a key focus in oncology drug discovery, both as a direct therapeutic target and a biomarker for guiding cancer treatment strategies.
KRAS inhibitors offer targeted cancer treatment, specifically addressing tumors with KRAS mutations, which are common in several types of cancers such as lung, colon, and pancreatic cancers. By specifically targeting mutated KRAS pathways, these inhibitors can effectively halt uncontrolled cell growth, potentially leading to improved treatment outcomes and prolonged survival in patients. Additionally, KRAS inhibitors can be integrated into personalized medicine strategies, providing more precise and effective treatment options based on individual genetic profiles.
Approach to the KRAS inhibitor development
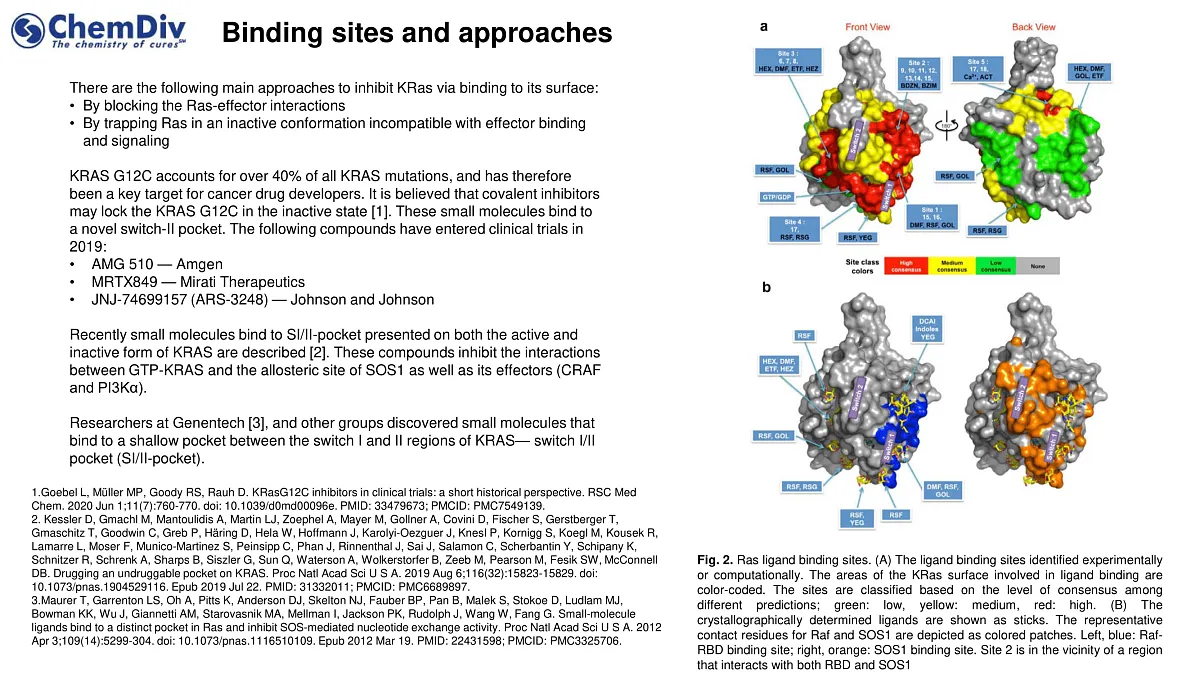
The main strategies to inhibit KRas involve targeting its surface through:
- Blocking the interactions between Ras and its effectors.
- Trapping Ras in an inactive conformation that prevents effector binding and signaling.
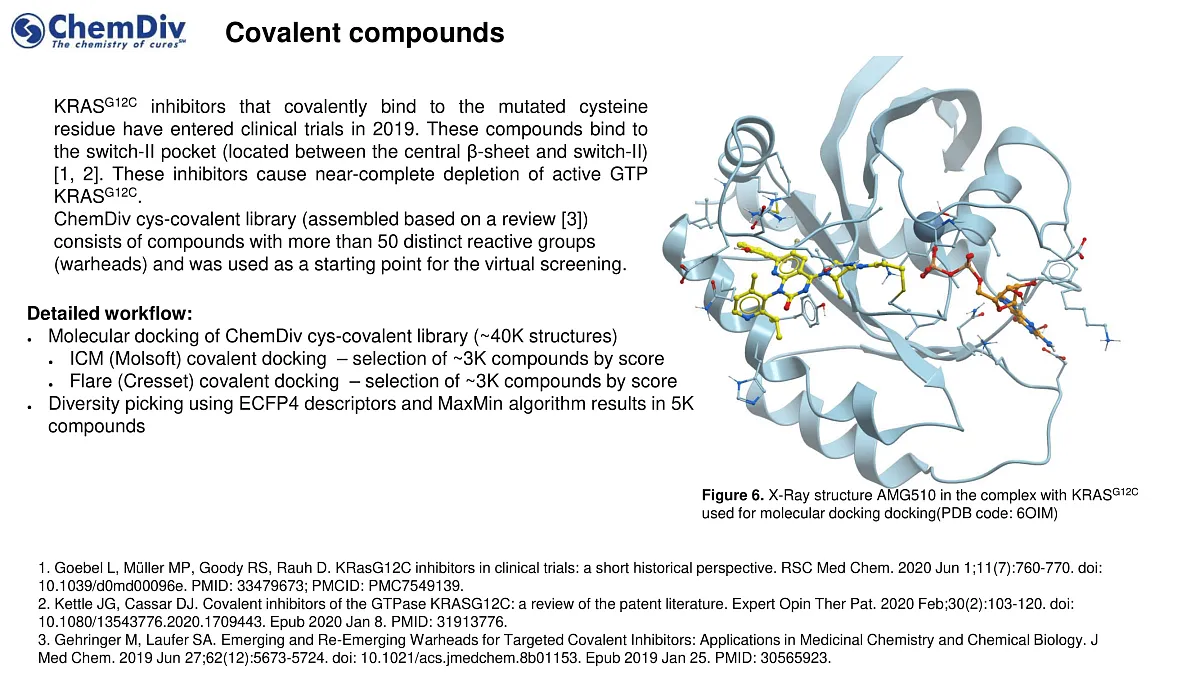
Covalent inhibitor molecules interact with a newly identified switch-II pocket. In 2019, several compounds entered clinical trials, including
- AMG 510 by Amgen,
- MRTX849 by Mirati Therapeutics,
- JNJ-74699157 (ARS-3248) by Johnson and Johnson.
Recent advancements have seen the development of small molecules that bind to the SI/II-pocket, present in both the active and inactive forms of KRAS. These compounds inhibit the interactions between GTP-bound KRAS and the allosteric site of SOS1, as well as its effectors such as CRAF and PI3Kα. Notably, researchers at Genentech and other groups have discovered molecules targeting a shallow pocket between the KRAS switch I and II regions, the SI/II-pocket.
Virtual screening strategy
The methodology integrates a consensus score and decision-making process based on a combination of sophisticated computational tools and theoretical knowledge, including the following stages:
- Standard 'MedChem' filters for assessing drug-likeness and chemical viability.
- Two-dimensional structure similarity analysis compared to known K/HRas ligands, utilizing the Tanimoto metric.
- The isosteric morphing procedure for modifying molecular structures while preserving their core properties.
- Identification and utilization of privileged structures or fragments known for their biological activity.
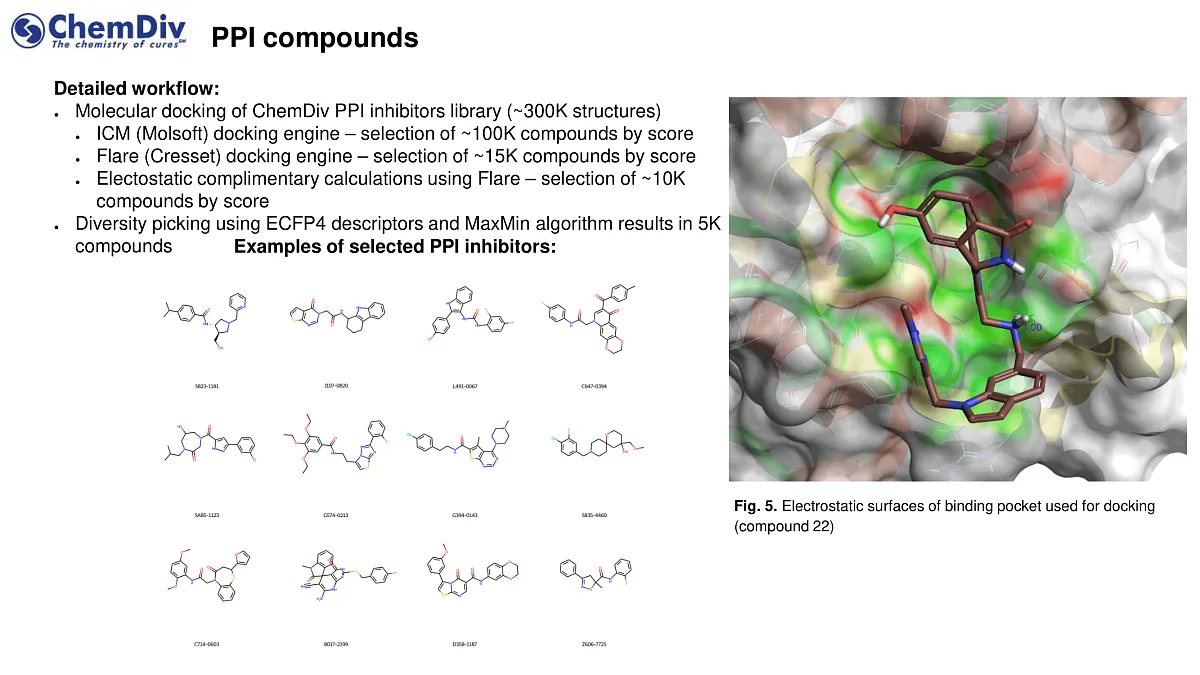
- A curated collection of approximately 5,000 compounds from a protein-protein interaction (PPI)-targeted library.
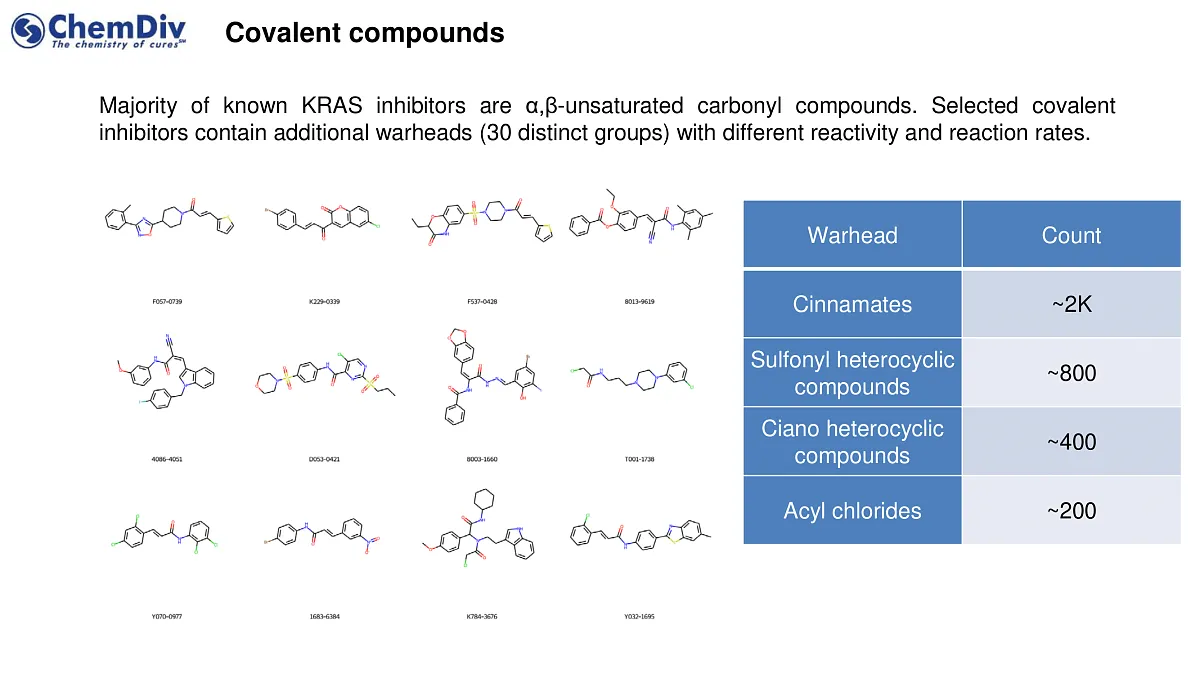
- A similarly sized set, around 5,000 compounds, featuring a covalent warhead specifically designed to target the G12C mutant in KRAS.
