cPLA2 inhibitors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cytosolic phospholipase A2 (cPLA2) is an enzyme that plays a crucial role in the metabolism of membrane phospholipids and the production of arachidonic acid, a precursor of various bioactive lipids like prostaglandins and leukotrienes. These metabolites are main components of inflammatory responses, making cPLA2 a potential target in the development of anti-inflammatory drugs. cPLA2 inhibitors play a vital role in drug discovery due to their potential in treating various inflammatory discorders. cPLA2, an enzyme crucial in the arachidonic acid pathway, is involved in the production of pro-inflammatory mediators like prostaglandins and leukotrienes. By inhibiting cPLA2, these drugs aim to reduce the synthesis of these mediators, thereby alleviating inflammation and associated symptoms. This makes cPLA2 inhibitors promising candidates for conditions such as asthma, rheumatoid arthritis, and other chronic inflammatory diseases. Furthermore, their potential is currently under investigation for neurodegenerative and cancer clinical practice, where inflammation is a key factor in disease progression.
ChemDiv’s library of cPLA2 inhibitors comprises more than 5K compounds.
The library was designed in accordance with the following strategies:
- Analysis of the cPLA2 inhibitor data in the ChEMBL database containing 399 ligands as a source of the primary scaffolds for developing the ligand-based virtual screening sets;
- Structure-based design utilizing human cPLA2 crystal structure.
The abovementioned strategy was implemented in the library build-up through the workflow:
- Exclusion of compounds with undesirable phys-chem characteristics:
- Pan-Assay Interference Compounds (PAINs) removal through substructure filtering;
- Non-MedChem-friendly SMARTS based filtering;
- Exclusion of highly lipophilic compounds (cLogP predicted by Molsoft software).
- Virtual screening procedure employing combined ligand-based methods and molecular docking;
- Structural diversity-driven compound selection using “Min-Max” algorithm.
Ligand development approach:
- Stock collection filtering on lipophilicity values (cLogP) predicted by ICM models
- Initial classification machine learning (ML) model-driven virtual screening filtering:
- The ChEMBL database, which contains 399 records featuring relevant data on the biological activity of known cPLA2 inhibitors, was utilized as a source for the training data. Decoys in the training set were generated using the DUD-E methodology, while those in the test set were collated from ChEMBL data
- Two independent ML models were deployed: a random forest model built on count-based Morgan fingerprints and an XGBoost model built on ECFP4 fingerprints.
- The compounds predicted by these models, along with their associated scores, were compiled as the final outcomes.
- Diversity-targeted compound selection for enhanced chemical space coverage
- Ligand-based selection from the stock collection yielded approximately 100,000 diverse compounds, fit for subsequent structure-based studies.
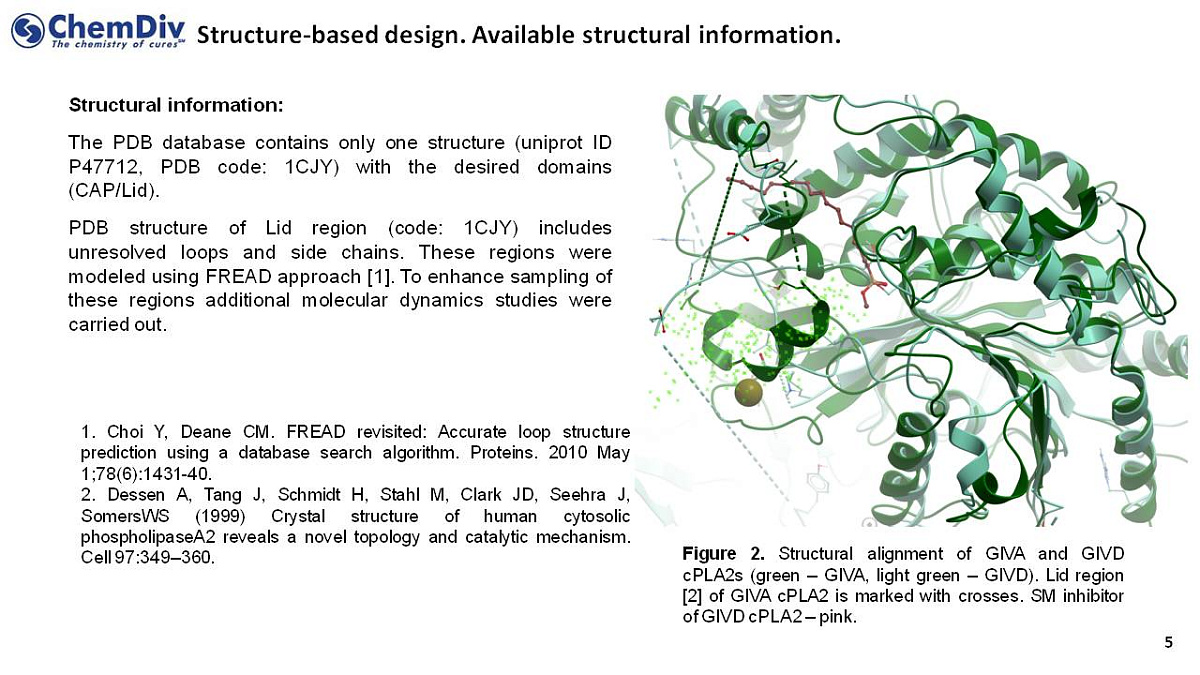
Structure-based studies were performed using the data obtained from the protein data bank (PDB), which provides one unique structure (uniprot ID P47712, PDB code: 1CJY) with the desired domains CAP/Lid. Lid region structure (PDB code: 1CJY) includes some unresolved loops and side chains serving as potential drug binding sites. The FREAD method was used to model those fragments, and then more molecular dynamics studies were done to improve the sampling of the target areas.
The key steps of that process include:
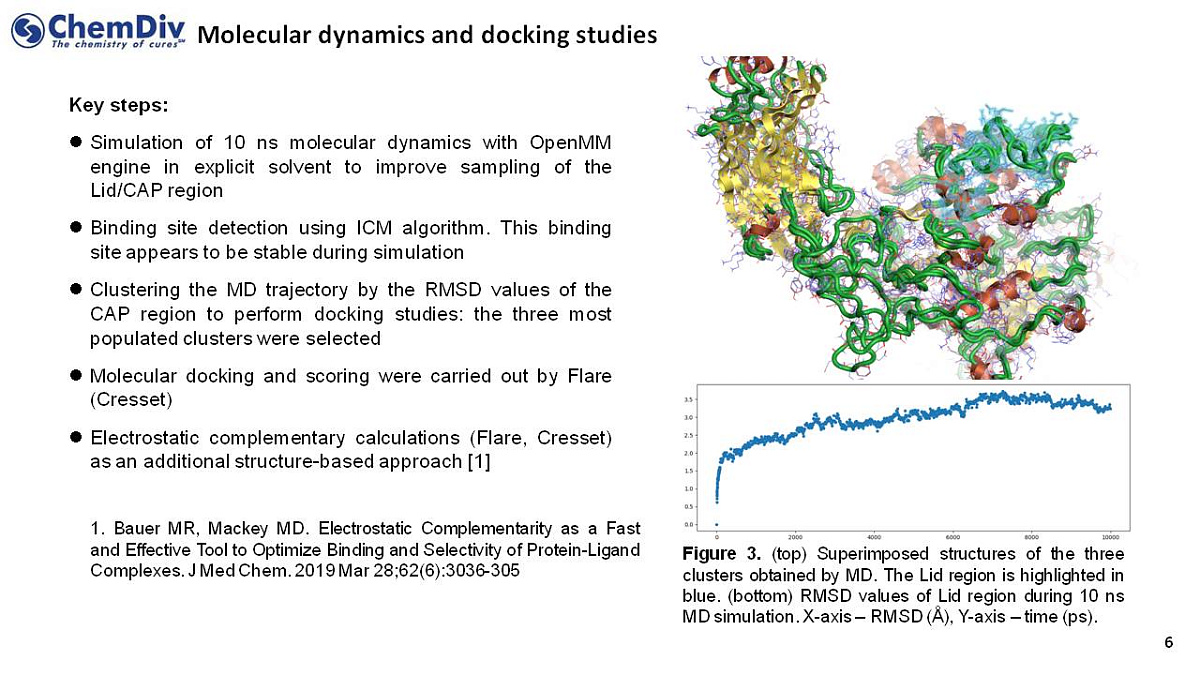
- A 10 ns molecular dynamics simulation was conducted using the OpenMM engine in explicitly modeled solvent molecules to improve sampling of the Lid/CAP regions;
- Binding site detection using ICM algorithm focusing on those appearing stable in simulation;
- Clustering the MD trajectories by the RMSD values of the CAP region utilized in the docking studies resulted in three most populated clusters selected;
- Molecular docking and scoring were conducted using the Flare software (Cresset);
- Complementary electrostatic calculations (Flare, Cresset) were implemented as additional structure-based methods.
The following criteria of the structure-based strategy were met:
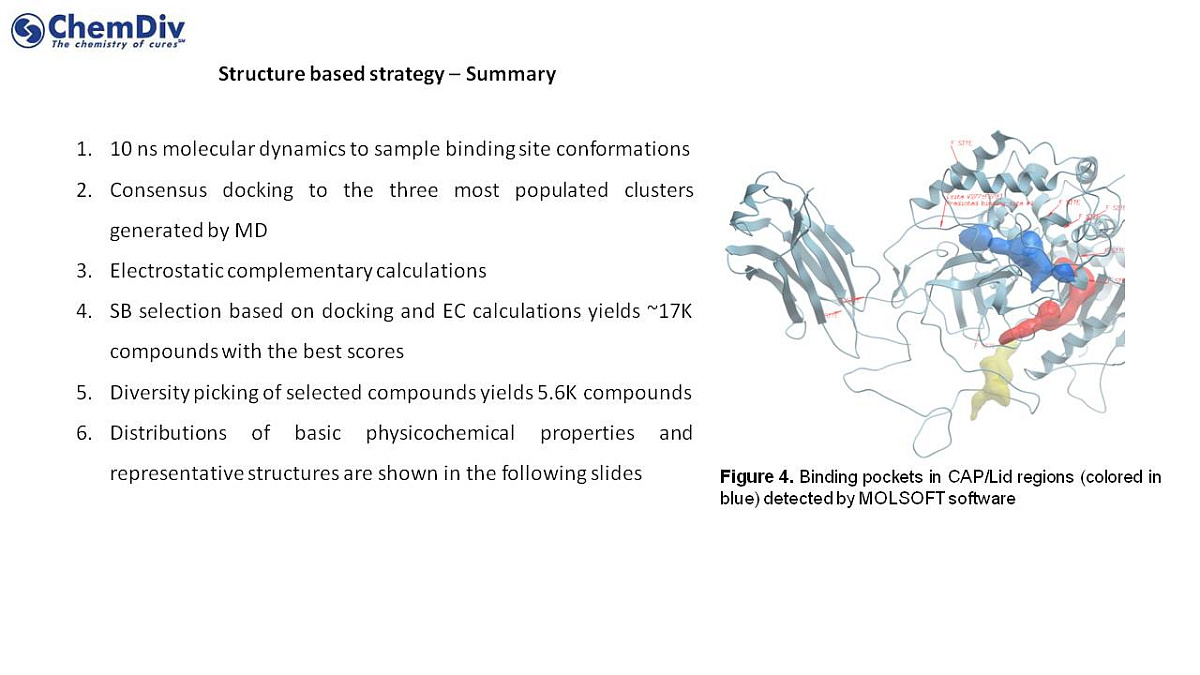
- 10 ns molecular dynamics deployed to sample the binding site conformations
- A consensus docking used to select three most populated clusters generated by MD
- Complementary electrostatic calculations
- Structure-based selection based on docking and EC calculations yields ~17K compounds with highest scores
- Diversity-based compound selection yields 5.6K compounds
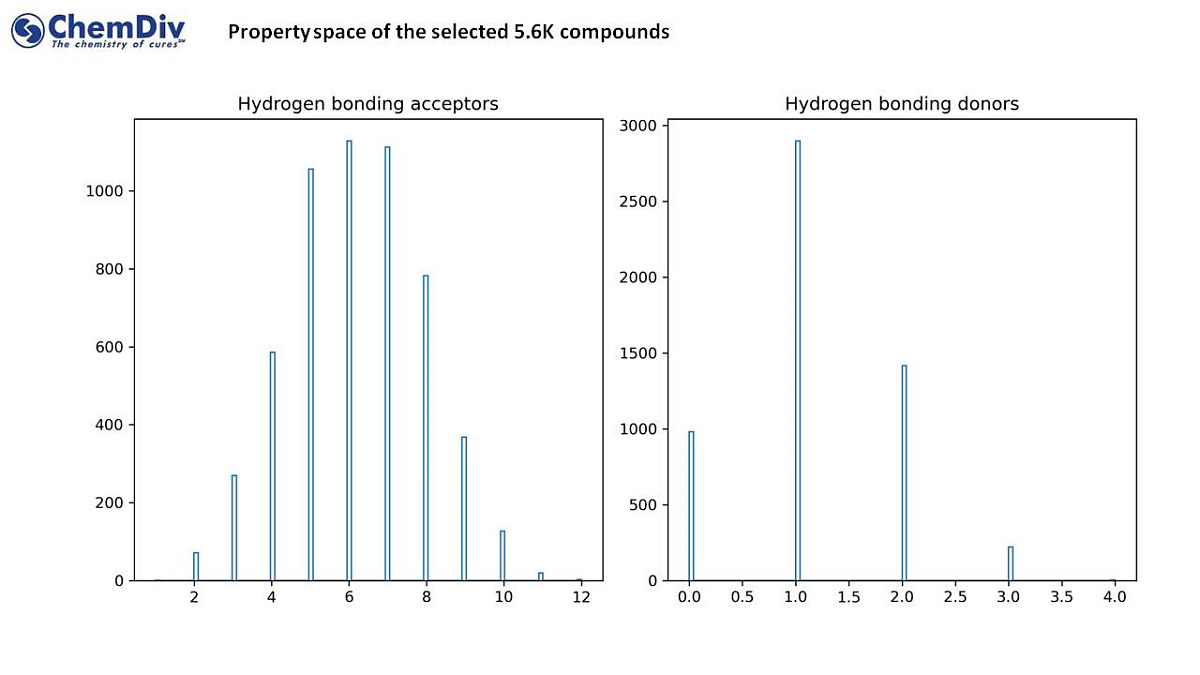
- Distributions of the basic physicochemical properties and representative structures are shown in the slide deck above.