PI3K-Targeted Library

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
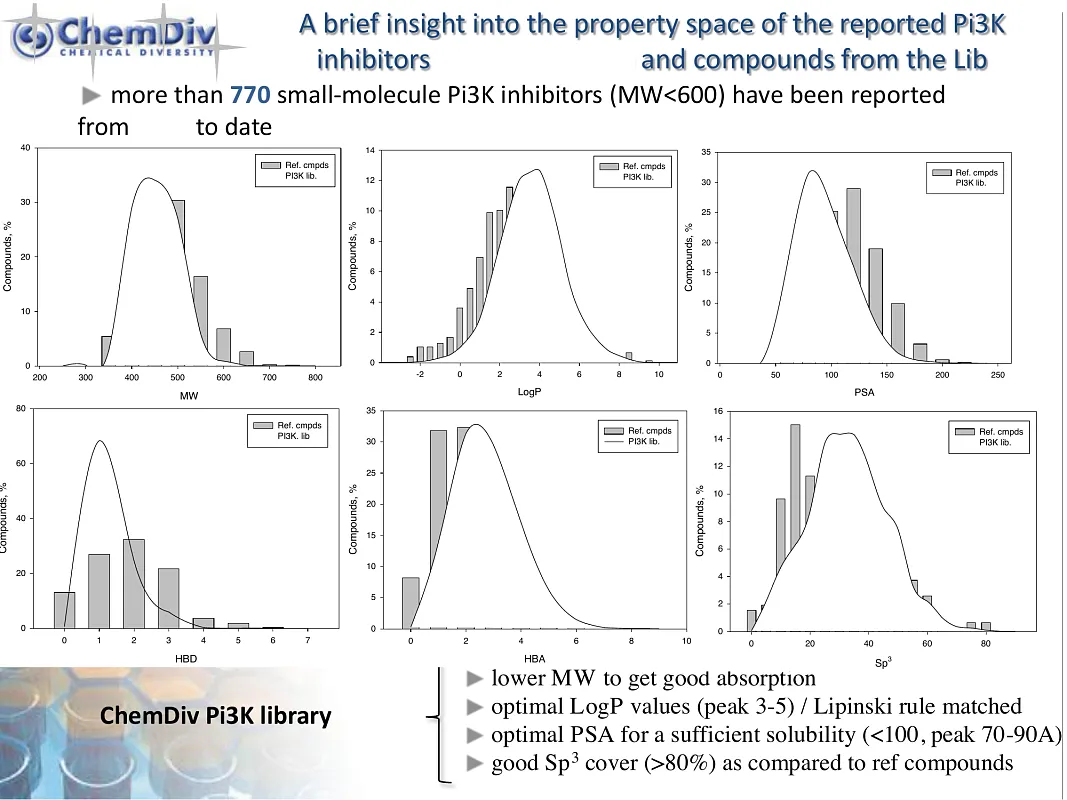
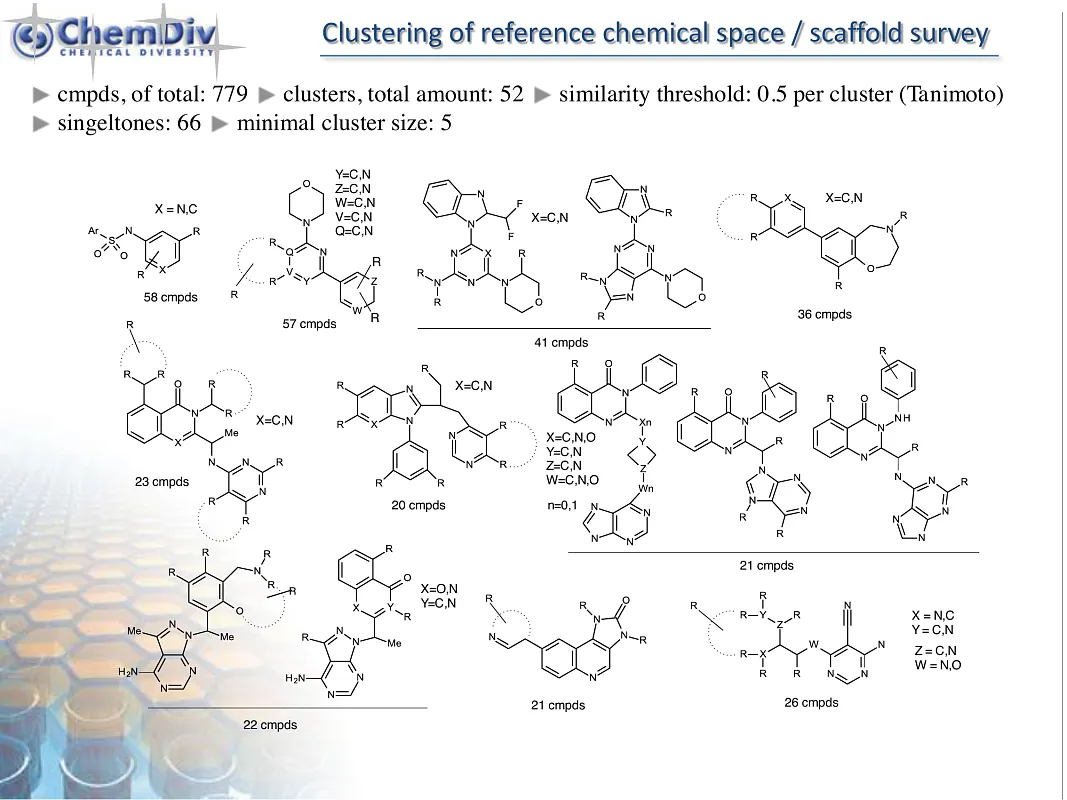
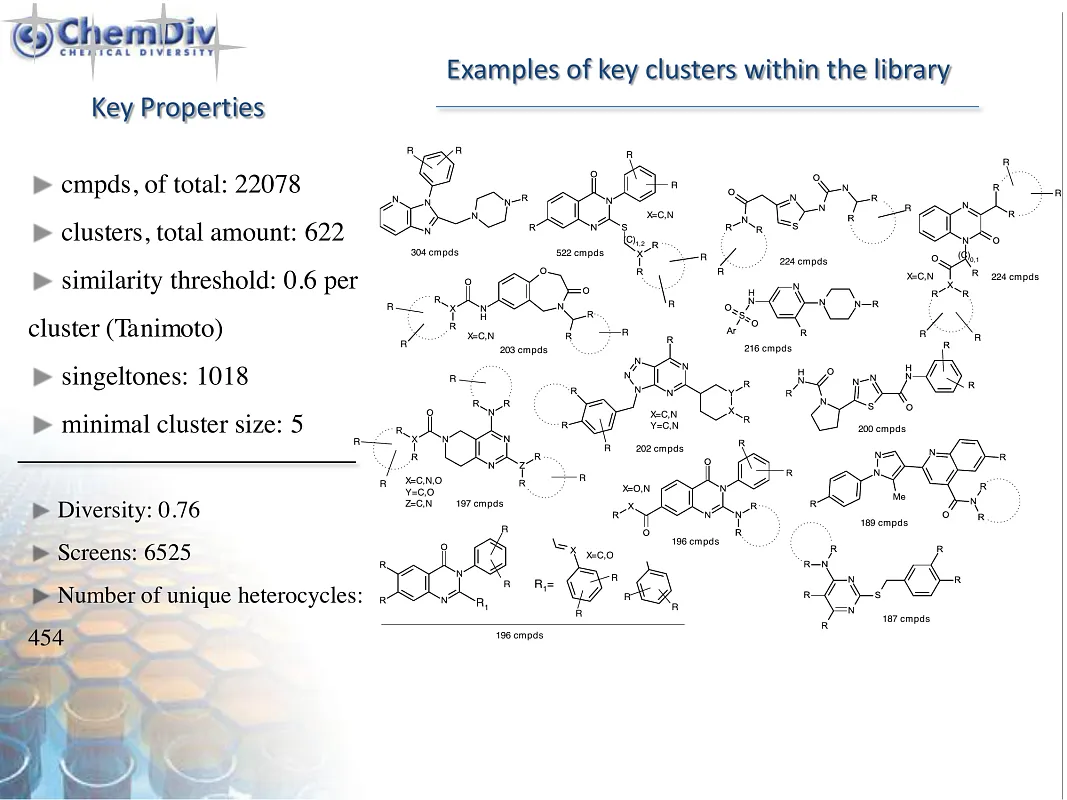
ChemDiv’s PI3K-Targeted Library contains 19,000 compounds.
The phosphatidylinositol-3-kinase (PI3K)/AKT/mammalian protein of rapamycin (mTOR) signaling pathway is a key enzyme involved in cellular processes such as cell motility, survival, metabolism, growth, and angiogenesis. Its aberrant activation often triggers tumor development and resistance to anticancer therapies, highlighting its critical role in almost all human cancers. This underscores the therapeutic potential of targeting the PI3K/AKT/mTOR pathway in cancer treatment. Inhibitors of PI3K, including PI3K/mTOR inhibitors, pan-PI3K inhibitors, and isoform-selective PI3K inhibitors, have demonstrated their ability to reduce cellular proliferation and induce apoptosis. The efficacy and safety of these inhibitors have been extensively studied in a variety of preclinical and clinical trials, revealing their significant potential in inhibiting tumor growth and slowing down its progression across the tissues [1].
PI3K inhibitors represent a significant advancement in the field of oncology drug discovery, offering a new avenue for targeting key intracellular signaling pathways implicated in cancer. Their ability to selectively inhibit the PI3K/AKT/mTOR pathway addresses a crucial need for more effective and safe treatments against various cancer types, especially those resistant to standard therapies. The development of these inhibitors has opened up promising possibilities for personalized medicine as researchers aim to personify therapeutic regimens based on specific genetic and molecular characteristics of different cancers.
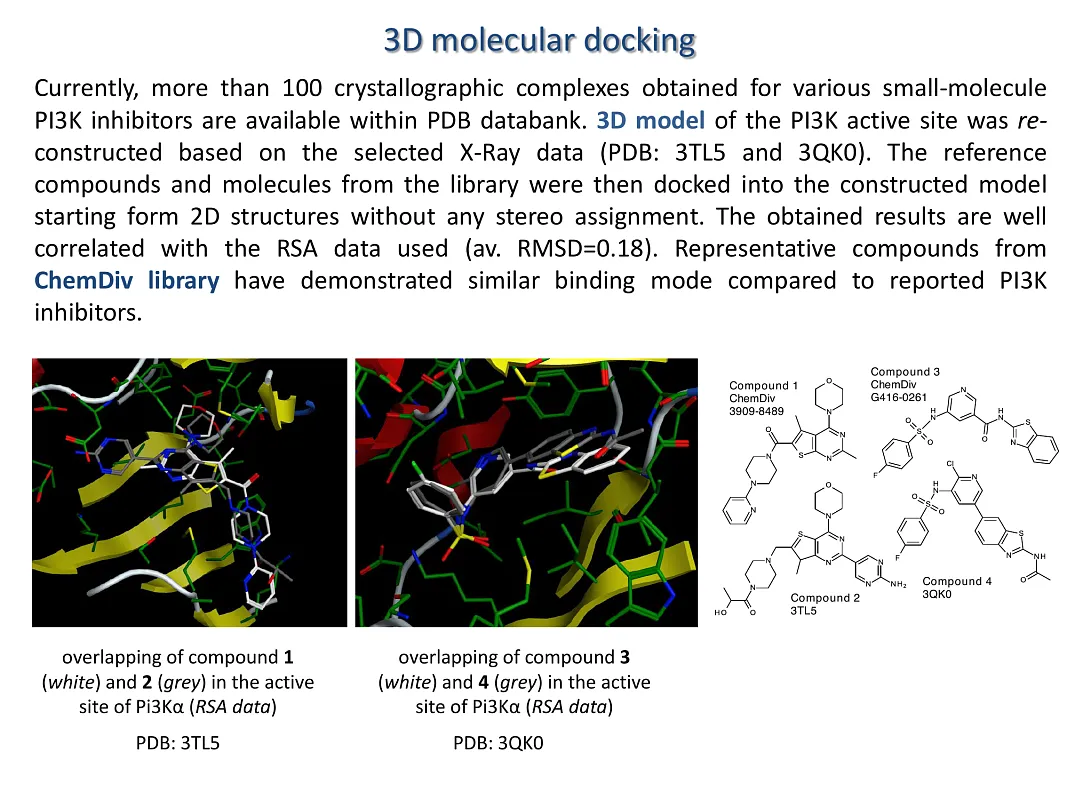
Currently, the PDB database contains over 100 crystallographic complexes of various small-molecule PI3K inhibitors. We reconstructed a 3D model of the PI3K active site based on selected X-ray crystallographic data (PDB codes: 3TL5 and 3QK0). Reference compounds, along with molecules from the library, were subsequently docked into this model, starting from their 2D structures with no stereochemical assignment. The docking results showed good correlation with the reference SAR data used, exhibiting an average RMSD of 0.18. Representative compounds from the ChemDiv library have demonstrated a binding mode similar to that of known PI3K inhibitors with a high level of affinity and selectivity.
References:
[1] J. Yang, J. Nie, X. Ma, Y. Wei, Y. Peng, and X. Wei, “Targeting PI3K in cancer: Mechanisms and advances in clinical trials 06 Biological Sciences 0601 Biochemistry and Cell Biology,” Mol. Cancer, 2019, vol. 18, no. 1, pp. 1–28, doi: 10.1186/s12943-019-0954-x.